一、标识标准
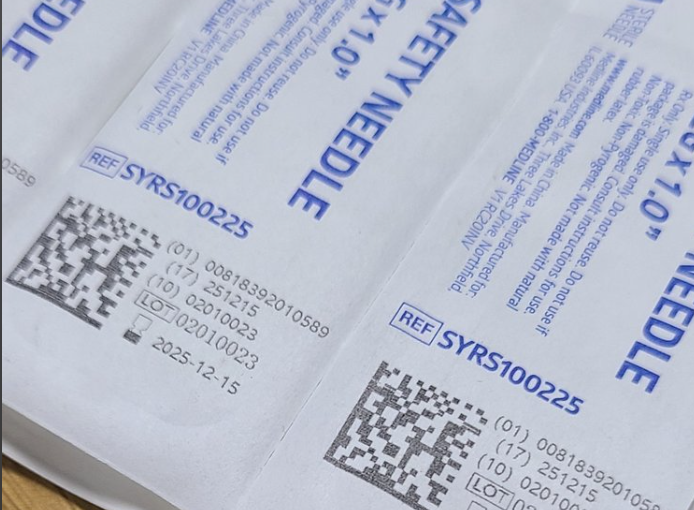
标签形式:医疗器械上需要采用机器可读和人类可读的标签形式,以确保信息可以便捷地获取和识别。
标签内容:包括制造商信息、产品型号、批次号、生产日期等,这些信息用于对医疗器械进行唯一标识和追踪。
二、数据格式
欧盟对医疗器械UDI的数据格式有规定,UDI编码需要采用特定的数据格式,以确保统一和一致。这有助于监管部门和制造商之间的信息交流和数据管理。
三、数据库连接
欧盟设立了UDI数据库,用于收集、管理和维护所有医疗器械的UDI编码信息。
制造商需要将医疗器械的UDI编码信息上传到UDI数据库,保证数据库中的信息和标签上的编码一致,有助于实现医疗器械的全生命周期追踪和管理。
四、标准协调
欧盟通过制定标准协调医疗器械UDI的实施。
这些标准包括技术标准、数据标准和业务流程标准等,用于统一和规范UDI的实施和管理,有助于提高UDI的标识一致性和信息互通性。
五、实施时间
根据欧洲医疗器械规则(MDR)和欧洲诊断医疗器械规则(IVDR),高风险和紧急使用的医疗器械首先实施UDI制度,然后逐步扩展至其他类别的器械。这意味着不同类别的器械在实施UDI制度时有不同的时间安排。
例如2022年5月26日,欧盟开始实施UDI,范围覆盖MDR的Ⅲ类产品;截至2023年5月26日,实施范围覆盖MDR的Ⅱa、Ⅱb、Ⅲ类 - 可重复使用产品、IVDR的ClassD产品。
六、制造商责任
制造商对医疗器械UDI的实施负有主要责任。
制造商需要确保医疗器械获得有效的UDI编号,并将相关信息上传到UDI数据库,还需要确保医疗器械上的标签符合欧盟的标准要求。
七、适用范围
欧盟对医疗器械UDI的实施范围涵盖了所有类别的医疗器械和体外诊断医疗器械。不论是进口的医疗器械还是在欧盟境内生产的医疗器械,都需要符合UDI的标准要求。